| Dictionnaire médical |
Abréviations S Sac Sacc Sacchar Sacr Sain Salm Salmonell Salp Salping San Sana Sang Sani Saph Sarc Scap Scapul Scar Schi Schist Schiz Scia Scie Science Scin Scint Scis Sclé Sclér Scol Scolio Scop Scot Seb Sebo Segm Segment Sept Septic Shif Sial Sibu Sidé Sidéro Sigmoïd Sign Signe de Sinu Sinus Siro Siru Sixi Soci Sodi Solu Solv Soma Somato Somi Soph Soud Sous Span Spas Spasm Spec Sper Sperm Sphi Sphinctér Sphy Sphygm Sphè Sphé Spin Spir Spiro Spla Splanchno Splé Splén Spon Spondyl Stad Stag Stap Staphyl Stapéd Stas Stat Sten Ster Sterco Stom Stre Strepto Stro Stéa Stéat Stén Sténo Stér Stéril Stérol Stéroïd Stéréo Sub Subi Subl Subs Suc Sudo Sudor Sueu Sueur Sui Suic Sulc Suns Supe Supr Supra Sur Sura Surc Surd Surf Suri Surj Surn Suro Surp Surr Surs Sus Sym Symb Symp Sympath Syn Syna Sync Synd Syndesm Syndrome Syne Syno Synovi Synt Syné Syph Syphil Syri Syring Syst Séba Sébo Sémi Sémio Sémé Séne Sér Séro Séru
| Syndrome de Wiskott-Aldrich | |
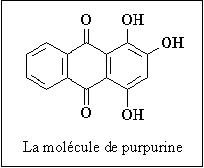
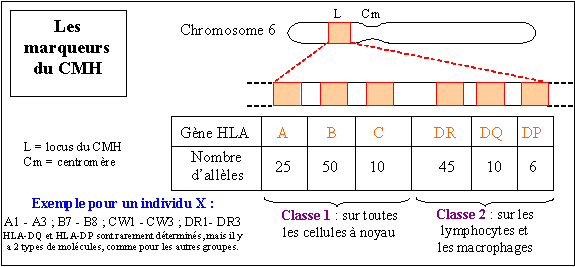
| Génétique, immunologie, dermatologie, pneumologie N. m. * syndrome : du grec sundromê , concours, réunion ; ensemble des symptômes qui caractérisent une maladie ; * Wiskott Alfred, pédiatre allemand né le 4 mars 1898 à Essen, mort en 1978 ; * Aldrich Robert Anderson, pédiatre américain né en 1917, mort en 1998. Le syndrome de Wiskott-Aldrich est aussi connu sous les noms de maladie de Werlhof familiale congénitale, purpura thrombocytopénique secondaire, syndrome d'Aldrich. L'un des principaux symptômes de cette maladie est un taux anormalement bas de plaquettes ou thrombocytes, ce qui lui a valu son autre nom de purpura thrombocytopénique (nombre de thrombocytes inférieur à la normale). Le gène responsable (gène WAS - abréviation de Wiskott-Aldrich Syndrome) est situé sur le chromosome sexuel X et la transmission se fait selon le mode récessif, c'est-à-dire que l'enfant doit recevoir le gène muté de chacun de ses parents pour développer la maladie, qui n'atteint que les garçons. Ce gène code la protéine WASP (Wiskott-Aldrich Syndrome Protein) qui joue un rôle essentiel dans la polymérisation de l'actine et dans la structure du cytosquelette ; si le gène est muté, la protéine est anormale. L'étiologie de ce syndrome semble assez complexe car les patients qui en sont atteints présentent aussi de nombreux dysfonctionnements de leur système immunitaire : les globulines E ou IgE (Ig = immunoglobuline) montrent un taux sanguin très supérieur à la normale, alors que les gammaglobulines M (ou IgM) sont très insuffisantes. Quant aux symptômes de ce syndrome, ils sont particulièrement nombreux. Parmi les plus significatifs : * apparition d'un purpura cutané (1) dès les premiers jours de la vie, d'hémorragies nasales, digestives, urinaires, d'un eczéma (2) avec violentes démangeaisons, au visage et aux plis de flexion, des infections récidivantes (otites, sinusites, furoncles, méningites, septicémie ...). Le pronostic de ce syndrome n'est toujours pas bon, même si des traitements peuvent être entrepris, essentiellement pour traiter les conséquences, comme des transfusions de plaquettes, des traitements pour l'eczéma, des antibiotiques pour les infections, entre autres. Récemment, des traitements par allogreffe de moelle osseuse par donneur HLA (3) compatible (4) ont été entrepris comme traitement de fond pour pallier la thrombocytopénie. A noter que le dépistage anténatal (avant la naissance de l'enfant) peut se faire pendant le premier trimestre de la grossesse. (1) Purpura : * du latin purpura, pourpre, relatif à des éruptions de taches rougeâtres sur la peau. Le purpura désigne une éruption cutanée plus ou moins importante de taches rougeâtres ou violacées, qui ont la particularité de ne pas disparaître à la vitropression (on appuie sur la lésion avec un verre de montre), ce qui permet de ne pas les confondre avec un érythème (rougeur cutanée). Elles sont dues à une extravasation sous-cutanée, c'est-à-dire à la rupture des capillaires sanguins et au passage du sang dans les tissus dermiques environnants. Il existe de nombreuses formes de purpura, car il ne s'agit en fait que de la manifestation d'autres pathologies sous-jacentes. Une NFS (numération formule sanguine) pratiquée par un laboratoire d'analyses biologiques et médicales, permet généralement de distinguer 2 grandes formes de purpura : * le purpura plaquettaire : comme son nom l'indique, il résulte d'une thrombopathie (les thrombocytes sont les plaquettes) de type thrombasthénie : fonctionnement anormal des plaquettes, mais aussi d'une insuffisance de plaquettes ou d'un purpura hémorragique héréditaire. Il peut s'agir soit d'une insuffisance de fabrication de ces thrombocytes dans la moelle osseuse, soit de leur destruction prématurée dans le sang, éventuellement par le système immunitaire. * Le purpura vasculaire, caractérisé essentiellement par une dégénérescence de la paroi des capillaires sanguins et qui se manifeste dans plusieurs pathologies: dermites ocres, vascularites, purpura rhumatoïde infantile ou syndrome de Schoenlein-Henoch, purpura de Bateman (ou purpura sénile de Bateman) qui apparaît sous forme de taches brunâtres chez les personnes âgées. Purpurique : adj. relatif au purpura. Purpurine : connue aussi sous le nom d'uroérythrine, la purpurine est un colorant rouge que l'on extrait naturellement de la garance (Rubia tinctorium). La purpurinurie correspond à la présence d'uroérythrine dans l'urine. (2) Eczéma : * du grec ekzein, bouillonner, bouillant. L'eczéma est une affection cutanée (de la peau) inflammatoire, qui peut être aiguë ou chronique. Il existe en fait une grande variété d'eczémas car les causes sont multiples et les manifestations nombreuses. Parmi les plus fréquentes, on retrouve l'érythème (rougeur) la formation de vésicules, petites "ampoules" remplies de liquide séreux, la lichénification avec formation de squames, petites peaux qui se détachent etc. Il existe actuellement toute une gamme de dermocorticoïdes (pommades, gels ou solutions contenant des dérivés de la cortisone) pour traiter avec plus ou moins de réussite ces formes eczémateuses. (3) HLA : Human Leucocyte Antigen. * HLA peut se traduire en français par "antigènes d'histocompatibilité" ; * anti : du préfixe anti- indiquant lhostilité, lopposition ou la défense (contre) ; * gène : du latin et du grec genesis [-gène, -genèse, -génie, -génique, -génisme, -génétique], naissance, formation, qui engendre ; * histo : du grec histos ou histion [hist(o), histio-], tissu ; * compatibilité : du latin compati [-compatible, -compatibilité], souffrir avec. Immunologie - N. m. * système : du grec sustêma [système], assemblage ; * HLA : abréviation de Human Leucocytes Antigens. Découvert en 1958 par Jean DAUSSET et connu aussi sous le nom de complexe majeur d'histocompatibilité, le système HLA est un ensemble de six gènes principaux, nommés A, B, C, DR (anciennement D1), DQ (anc. D2) et DP (anc. D3) et présents sur le bras court du chromosome 6. Les gènes A, B et C sont qualifiés de gènes de classe I qui codent les molécules du CMH-I, alors que les régions DR, DQ et DP contiennent 23 à 25 gènes dits de classe II qui codent les molécules du CMH-II. Depuis le séquençage des gènes du CMH humain, on connaît 128 gènes actifs pour le complexe HLA. Ces gènes codent des glycoprotéines qui se trouvent à la surface de toutes nos cellules à noyau et qui jouent un rôle déterminant dans la reconnaissance su soi modifié et dans le rejet éventuel d'une greffe. Ils codent également d'autres molécules, comme des enzymes, certains éléments du complément, des molécules qui interviennent dans la communication entre cellules immunitaires, entre autres. Les individus étant généralement hétérozygotes pour ces gènes (compte tenu du nombre important d'allèles pour chacun d'eux), il y a donc douze sortes de glycoprotéines à la surface de nos cellules, avec une probabilité de 1/4 que deux enfants issus de mêmes parents aient le même CMH. Le schéma suivant sonne, pour les principaux gènes, le nombre d'allèles connus en 1992, et qui ne cesse d'augmenter. Les glycoprotéines ou antigènes membranaires sont formées de deux chaînes . Leur structure quaternaire (disposition dans l'espace) est connue pour la plupart d'entre elles. Les molécules HLA que l'on trouve à la surface de la majorité des cellules comportent une chaîne invariable : ß2 microglobuline, codée par un gène, qui ne fait pas partie du CMH, et présent sur le chromosome 15. Cette chaîne β2 est insérée dans la membrane et liée de façon non covalente à une chaîne lourde a polymorphique située à l'extérieur de la cellule, codée par les gènes A, B et C et qui présente une cavité dans laquelle peuvent venir s'insérer de petits peptides d'environ 9 acides aminés. Les molécules HLA sont formées de deux chaînes polymorphiques (a, b) comportant chacune deux domaines extra-membranaires présentant aussi une cavité extérieure à la cellule et pouvant contenir de petits peptides. Une chaîne légère invariable (li) codée par un gène situé sur le chromosome 5, est associée aux deux chaînes précédentes sur la face interne de la membrane. Quant à la chaîne β, elle est codée par les gènes des régions D. Les molécules HLA du premier groupe, donc de classe I, sont supposées être responsables de la présentation des peptides (9 aa) issus de la dégradation permanente par chaque cellule de ses propres protéines (protéines endogènes). La cellule présenterait ainsi en surface en permanence des fragments des protéines qu'elle synthétise. Ces protéines seraient bien sûr modifiées dans le cas d'une infection virale, la cellule synthétisant alors les protéines virales. Les molécules HLA du second groupe, donc de classe II, sont supposées présenter elles aussi des peptides de 9 aa, mais qui sont issus du milieu extérieur, ont été absorbés par la cellule par endocytose, puis dégradés. Il semblerait qu'il existe un seuil à partir duquel la présentation des peptides provoque une réaction immunitaire. Ce seuil correspond à environ de 200 molécules HLA associées au même peptide. La plupart des peptides présentés par les molécules HLA sont des fragments de ces mêmes molécules HLA. De nombreuses séquences de peptides sont identiques (homologues) à celles de peptides de micro-organismes. Enfin, si le nombre théorique de peptides possible est de 5.1011 (9 aa parmi 20 aa) , il semble que le nombre réel soit beaucoup plus restreint du fait de leur aptitude ou non à établir des liaisons avec les molécules HLA. Sur les hématies (cellules sans noyau) se trouvent des antigènes qui déterminent les groupes sanguins et qui déterminent le complexe mineur d'histocompatibilité.
© Georges Dolisi | |
| < vers le menu principal | Politique de confidentialité | App Privacy Policy | ^ haut de la page |
| © 2006, 2020 Medicopedia | Powered by JIMaroc |