| Dictionnaire médical |
Abréviations S Sac Sacc Sacchar Sacr Sain Salm Salmonell Salp Salping San Sana Sang Sani Saph Sarc Scap Scapul Scar Schi Schist Schiz Scia Scie Science Scin Scint Scis Sclé Sclér Scol Scolio Scop Scot Seb Sebo Segm Segment Sept Septic Shif Sial Sibu Sidé Sidéro Sigmoïd Sign Signe de Sinu Sinus Siro Siru Sixi Soci Sodi Solu Solv Soma Somato Somi Soph Soud Sous Span Spas Spasm Spec Sper Sperm Sphi Sphinctér Sphy Sphygm Sphè Sphé Spin Spir Spiro Spla Splanchno Splé Splén Spon Spondyl Stad Stag Stap Staphyl Stapéd Stas Stat Sten Ster Sterco Stom Stre Strepto Stro Stéa Stéat Stén Sténo Stér Stéril Stérol Stéroïd Stéréo Sub Subi Subl Subs Suc Sudo Sudor Sueu Sueur Sui Suic Sulc Suns Supe Supr Supra Sur Sura Surc Surd Surf Suri Surj Surn Suro Surp Surr Surs Sus Sym Symb Symp Sympath Syn Syna Sync Synd Syndesm Syndrome Syne Syno Synovi Synt Syné Syph Syphil Syri Syring Syst Séba Sébo Sémi Sémio Sémé Séne Sér Séro Séru
| Syndrome de Tangier | |
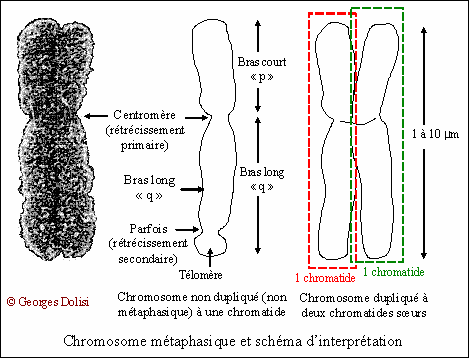
| Génétique, maladies orphelines, hépatogastroentérologie N. f. * Tangier : du nom d'une ville sur l'île "Tangier Island" dans la Baie de Chesapeake, au large de la Virginie ; * hypo : du grec hupo , dessous, indiquant aussi une qualité ou une intensité inférieures à la normale ; * alpha : première lettre de lalphabet grec (a), utilisée avec dautres lettres grecques pour classer des éléments en biochimie, ou pour désigner un rayonnement en physique ou en médecine nucléaire ; * lipo : du grec lipos, liparos {lip(o)-}, graisse, gras ; * protéine : du grec tardif prôteios, signifiant "qui occupe le premier rang" ou "de première qualité", lui-même dérivé de protos, premier, relatif aux protides ou aux protéines, composées essentiellement de C, H, O et N ; * émie : du grec haima, {-émie, héma-, hémat(o)-, hém(o)-} : relatif au sang. [Angl. : Tangier disease, analphalipoproteinemia, hypoalphalipoproteinemia] Syn. : hypoalphalipoprotéinémie, analphalipoprotéinémie, syndrome de Tangier. La maladie de Tangier est une maladie génétique rare ou maladie orpheline, puisqu'on ne connaît que moins de cinquante cas dans une vingtaine de familles dans le monde. Quelques explications pour mieux comprendre cette maladie du transport du cholestérol. * Sur le chromosome 9q31, c'est-à-dire sur le bras long (q) du 9e chromosome, à un emplacement ou locus précis nommé 31, se trouve le gène ABCA1 (ATP Binding Cassette Transporter). En fait, il s'agit de la 9e paire de chromosomes, et nous avons donc ce gène en double, chacun hérité de l'un de nos deux parents. ATP désigne l'adénosine triphosphate qui est le principal transporteur d'énergie dans toutes nos réactions cellulaires. Ce gène ABCA1 code la fabrication d'une protéine du même nom (ABCA1) qui est enchâssée dans la membrane de nos cellules et dont le rôle principal est de permettre à certaines molécules dont le cholestérol, de quitter l'intérieur de la cellule pour passer dans le milieu extracellulaire. Ces molécules sont trop grandes pour passer "naturellement" de l'intérieur vers l'extérieur et ce passage nécessite la présence dans la membrane de ces protéines particulières qui vont permettre ce transfert qualifié d'actif, en consommant de l'énergie, donc de l'ATP. Ce n'est qu'une fois dans le milieu extracellulaire que le cholestérol va se lier à l'apoA1 et aux phospholipides pour former le complexe HDL. * Chez un nombre très limité de personnes, l'un des chromosomes n° 9, plus rarement encore les deux, présente(nt) un gène ABCA1 qui a muté, c'est-à-dire qu'il ne fabrique plus la bonne protéine. Sans trop entrer dans les détails, on constate que dans un gène muté, la séquence des bases azotées qui composent la double hélice de l'ADN n'est plus la bonne. On comprend aisément ce qui se passe dans ce cas : le cholestérol ne peut plus quitter la cellule pour se rendre vers le foie et être éliminé. Il va s'accumuler préférentiellement dans des organes comme le foie et la rate, les artères, la cornée, les amygdales, entre autres. La maladie de Tangier résulte donc d'un dysfonctionnement du transport du cholestérol. * Une précision importante : Cette maladie se transmet selon le mode autosomique récessif. Autosomique : qualifie les chromosomes non sexuels, c'est-à-dire les 22 premières paires (la 23e paire étant conventionnellement celle des chromosomes sexuels, XX chez la femme, XY chez l'homme). Récessif : il faut que l'enfant reçoive de chacun de ses parents un chromosome avec le gène muté pour qu'il développe la maladie. Il est alors homozygote pour ce gène muté, puisqu'il l'a sur les deux chromosomes n° 9. S'il ne reçoit le gène muté que de l'un de ses parents, il est hétérozygote ou "porteur sain" et ne développe aucun symptôme de la maladie, car il produit suffisamment de protéines ABCA1 normales. * Principaux symptômes de la maladie de Tangier, sachant qu'aucun d'entre eux n'est obligatoire et qu'il existe des cas totalement asymptomatiques. ● Chez les nouveau-nés, les amygdales sont jaune-orangé du fait de l'accumulation d'esters du cholestérol et de taille supérieure à la normale. ● Hypolipidémie, en particulier baisse sévère du taux des HDL (high density lipoprotein, le "bon" cholestérol) qui est < 0,1 g/L, ainsi que de l'ApoA1. C'est ce qui justifie son autre nom d'hypoalphalipoprotéinémie ou d'analphalipoprotéinémie. ● Athérosclérose prématurée disséminée, avec atteinte coronarienne fréquente et risque accru d'infarctus du myocarde. ● Opacité cornéenne résultant de l'accumulation d'esters de cholestérol. ● Hépatomégalie et splénomégalie (augmentation anormale du volume du foie et de la rate). ● Sur la muqueuse du côlon sigmoïde et du rectum, on observe des dépôts jaunâtres de cholestérol, visibles en coloscopie sous forme de plaques de 2 à 3 mm. ● Les amygdales ne sont pas les seuls organes lymphoïdes touchés : on peut aussi observer de petites adénopathies inguinales (à l'aine), axillaires (aux aisselles) et cervicales. ● Le fait que le cholestérol ne puisse plus sortir des cellules va provoquer une cytolyse (destruction cellulaire) plus ou moins importante, ce qui se traduit par un taux d'ALAT supérieur à 50 UI/L (normale < 25). ● Parfois rétinite pigmentaire, légère hypertriglycéridémie, entre autres. * Traitements. Comme indiqué plus haut, il s'agit d'une maladie orpheline et il n'existe aujourd'hui aucun traitement, sinon symptomatique. Cela signifie que l'on soigne au coup par coup les symptômes qui deviennent gênants ou dangereux : angioplastie coronaire transluminale si des athéromes obstruent les artères coronaires, greffe de cornée pour éviter la cécité, amygdalectomie (ou tonsillectomie) chez les jeunes enfants, greffe du foie, entre autres. Les espoirs actuels reposent sur une thérapie génique qui n'est pas encore au point.
© Georges Dolisi | |
| < vers le menu principal | Politique de confidentialité | App Privacy Policy | ^ haut de la page |
| © 2006, 2020 Medicopedia | Powered by JIMaroc |