| Dictionnaire médical |
Abréviations S Sac Sacc Sacchar Sacr Sain Salm Salmonell Salp Salping San Sana Sang Sani Saph Sarc Scap Scapul Scar Schi Schist Schiz Scia Scie Science Scin Scint Scis Sclé Sclér Scol Scolio Scop Scot Seb Sebo Segm Segment Sept Septic Shif Sial Sibu Sidé Sidéro Sigmoïd Sign Signe de Sinu Sinus Siro Siru Sixi Soci Sodi Solu Solv Soma Somato Somi Soph Soud Sous Span Spas Spasm Spec Sper Sperm Sphi Sphinctér Sphy Sphygm Sphè Sphé Spin Spir Spiro Spla Splanchno Splé Splén Spon Spondyl Stad Stag Stap Staphyl Stapéd Stas Stat Sten Ster Sterco Stom Stre Strepto Stro Stéa Stéat Stén Sténo Stér Stéril Stérol Stéroïd Stéréo Sub Subi Subl Subs Suc Sudo Sudor Sueu Sueur Sui Suic Sulc Suns Supe Supr Supra Sur Sura Surc Surd Surf Suri Surj Surn Suro Surp Surr Surs Sus Sym Symb Symp Sympath Syn Syna Sync Synd Syndesm Syndrome Syne Syno Synovi Synt Syné Syph Syphil Syri Syring Syst Séba Sébo Sémi Sémio Sémé Séne Sér Séro Séru
| Sang | |
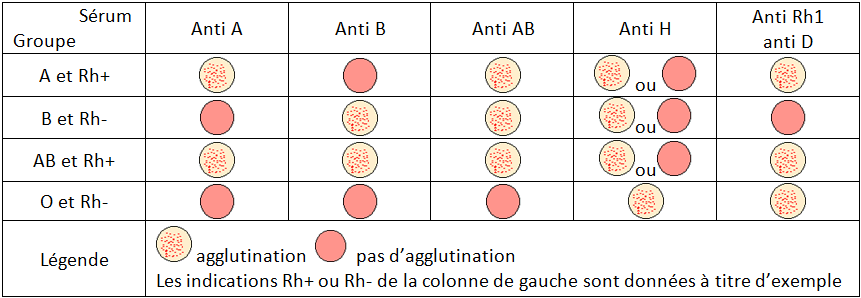
| Hématologie, médecine biologique N. m. {Angl. : Blood, total blood} * sang : du latin sanguis, sanguinis, sanguinare {sanguin(o)-, -sanguin, -sanguinolent}, liquide rouge qui circule dans les veines et les artères. Cette rubrique comprend 17 schémas. Pour des raisons techniques, seuls les 5 premiers sont placés ci-dessous. Les 5 suivants sont dans "Sang 1", les 5 suivants dans "Sang 2" et les 2 derniers dans "Sang 3". Le sang est un tissu liquide dont l'un des principaux rôles est d'assurer les transports gazeux, mais aussi des nutriments vers les cellules, des déchets cellulaires vers les organes d'épuration (reins, foie ...), des hormones et anticorps. Il est contenu dans un système circulatoire clos (artères et veines) mais des échanges de liquide (lymphe) existent au niveau des capillaires : c'est la transsudation. Les mouvements de la lymphe dans le corps (sortie des capillaires par transsudation, puis passage dans les espaces intercellulaires et pénétration dans les cellules) dépendent d'un nombre important de facteurs : pression oncotique (les protéines sanguines attirent le sérum des tissus vers l'intérieur des capillaires), pression hémodynamique (la pression sanguine pousse le sérum à travers les parois capillaires vers les espaces extracellulaires, pression osmotique, entre autres. C'est cette lymphe interstitielle qui assure les échanges cellulaires. I - Groupes sanguins PRINCIPE : Lorsqu'un corps étranger ou antigène (AG) est introduit dans notre organisme (organe non compatible, sang anisogroupe, bactérie, virus, pollen ...), notre système immunitaire fabrique un anticorps (AC) capable de le neutraliser. Les réactions sont complexes et mettent en jeu de nombreux éléments : polynucléaires, lymphocytes, complément ... En ce qui concerne le sang, les 4 groupes A, B, AB et O sont déterminés par la présence ou l'absence de 2 protéines agglutinogènes, jouant le rôle d'antigènes, à la surface des hématies et appelées A et B. Particularité : les AC existent de façon naturelle dans le sang. Ce sont les agglutinines a (anti A) et b (anti B). C'est ainsi qu'une personne du groupe A possède la protéine A sur ses hématies et possède également l'agglutinine b (anti B) dans son plasma ; une personne du groupe B possède la protéine B et l'agglutinine a (anti A) ; si vous êtes du groupe AB (receveur universel) vous avez à la fois la protéine A et la protéine B, mais ni l'agglutinine a, ni la b et pouvez de ce fait recevoir les 4 groupes en transfusion ; enfin, une personne du groupe O (donneur universel) n'a ni la protéine A, ni la B, ce qui explique qu'elle puisse donner son sang aux 4 groupes, mais possède les agglutinines a et b et ne peut donc recevoir que du sang du groupe O. Voir tableau. Exemple : si l'on transfuse du sang B à un sujet A, ses AC b vont agglutiner le sang B et atteindre du même coup le sang du receveur. C'est l'accident hémolytique (hémo = sang). Remarque : Si la quantité de sang à transfuser est importante, (supérieure à 1 litre), et à plus forte raison s'il s'agit d'une exsanguino-transfusion ou d'une circulation extracorporelle, on ne doit pas injecter de sang dont les AC agglutineraient ou revêtiraient les globules du receveur. La transfusion devient alors nécessairement isogroupe. FACTEUR RHÉSUS : même principe que les groupes sanguins, mais totalement indépendant de ces groupes. 85% des humains ont cette protéine (= agglutinogène) et sont Rh+ (DD ou Dd). 15% ne l'ont pas et sont Rh- (dd). On utilise un sérum anti Rh (ou anti D) pour la détermination. A noter cependant que les anticorps anti Rh n'existent pas de façon naturelle dans le sang. Remarque : sur les hématies de tous les groupes existe la substance H. Sur un groupe A "fort", (ou B "fort" ou AB "fort"), il y en a très peu => anti H - . Sur un groupe A "faible", il y en a plus => anti H +. Chez une personne O : ni A, ni B => uniquement H => anti H +. Anti H est + chez tous les O, mais n'est pas caractéristique de ce groupe. II - Coagulation ou hémostase : Les facteurs de la coagulation * facteur : du latin factor, de facere [facteur], faire ; * coagulation : du latin coagulare [-coagulant, -coagulation], donner à un liquide une consistance solide, le figer. A l'exception du facteur IV qui est le calcium (Ca++), les facteurs de coagulation sont tous des protéines, substances présentes dans le sang et qui participent à la coagulation sanguine ou hémostase. Toute anomalie ou dysfonctionnement d'un ou plusieurs de ces facteurs provoque une forme plus ou moins sévère d'hémophilie. On connaît actuellement 13 facteurs de coagulation qui sont notés par des chiffres romains. Pour mieux comprendre ce qui suit, petit rappel sur la coagulation. C'est un ensemble de mécanismes qui fait passer le sang de l'état liquide à un état de gel ou caillot, en transformant le fibrinogène soluble en fibrine insoluble qui forme l'armature du caillot. On distingue classiquement 3 étapes : 1) La thromboplastinoformation ou transformation de la prothrombinase selon 2 voies différentes : intrinsèque et extrinsèque, et qui aboutit à la formation d'une enzyme : le facteur X activé. 2) La thrombinoformation qui aboutit à la formation de thrombine sous l'action de la thrombinase. 3) La fibrinoformation qui permet, grâce à la thrombine, la transformation du fibrinogène en fibrine. * Facteur I : c'est le fibrinogène. Du latin et du grec genesis [-gène, -genèse, génique], naissance, formation, qui engendre. Synthétisé par le foie, le fibrinogène est une glycoprotéine qui existe à l'état soluble dans le sang circulant. Sous l'action d'une enzyme (la thrombine), le fibrinogène est transformé en fibrine insoluble (c'est la fibrino-formation) et joue donc un rôle majeur dans la coagulation du sang (hémostase). A l'électrophorèse, le fibrinogène migre entre les ß et les g globulines. Les valeurs plasmatiques normales sont comprises entre 3 à 5 g/L (ou 9 à 15 mmol/L) de fibrinogène par litre de sang. C'est la fibrinogénémie, alors que la fibrinémie représente le taux de fibrine du plasma. * Facteur II : la prothrombine. Du grec thrombos [thromb(o)-], caillot, en rapport avec la coagulation du sang. C'est une substance qui, sous l'influence d'une dizaine d'autres facteurs, fait coaguler le sang par polymérisation du fibrinogène soluble en fibrine insoluble. La mesure de la prothrombine est très importante chez les personnes qui suivent un traitement par anticoagulants. On pratique essentiellement 2 examens : 1 - Le taux de prothrombine permet de tester le bon fonctionnement de certains facteurs de la coagulation. Ils sont abaissés en cas de maladie hépatique et lorsqu'un malade prend des médicaments anticoagulants par voie orale. C'est l'examen de choix pour contrôler l'efficacité d'un traitement. Ce taux de prothrombine ou "TP", doit alors être strictement compris entre 25% et 35%, alors qu'il est normalement supérieur à 70%. S'il est inférieur à 25%, il y a risque d'hémorragie, et s'il est supérieur à 35%, le traitement n'est plus suffisamment efficace. 2 - Le temps de prothrombine, appelé également temps de Quick, est le temps que met le plasma sanguin (partie liquidienne du sang) à coaguler quand on le met en présence dun extrait de tissu humain, animal ou synthétique appelé thromboplastine, cytozyme ou thrombokinase. Il sagit dun ensemble denzymes nécessaires à la coagulation du sang et permettant de transformer la prothrombine en thrombine. La mesure du temps de Quick est faite à partir dun prélèvement de sang du malade. Ce test sert à savoir si le patient présente une tendance à faire des hémorragies à la suite dune carence en certains facteurs normalement présents dans le sang : facteur II, V, VII, X qui interviennent dans la coagulation normale du sang. Le temps de prothrombine sexprime sous la forme dun indice INR (International Normalized Ratio ou rapport international normalisé). Un temps de prothrombine anormal indique un problème de coagulation, dorigine soit pathologique soit thérapeutique. Une atteinte du foie ou un manque de vitamine K peuvent être à lorigine de perturbations du temps de prothrombine. Le TP sert à surveiller les traitements anticoagulants par les antivitamines K. On appelle facteur IIa la thrombine. * Facteur III ou facteur tissulaire : la thromboplastine tissulaire ou thrombokinase (les autres sont des facteurs plasmatiques). Ce facteur III de la coagulation est présent à la surface des vaisseaux sanguins. Sa quantité à la surface augmente selon certains signaux comme l'inflammation. Elle provoque l'activation du facteur VII de la coagulation - voir ci-dessous. * Facteur IV : le calcium. Du latin calx, calcis, chaux. Métal de symbole chimique Ca. C'est l'élément N° 20 de masse atomique Ca = 40,08 et de densité 1,54. Il s'oxyde à l'air en formant la chaux vive CaO. Il participe à un grand nombre de phénomènes physiologiques : synthèse du tissu osseux et dentaire, coagulation du sang, conduction nerveuse, contraction musculaire. L'hypocalcémie provoque des convulsions et des tremblements (spasmophilie, tétanie). * Facteur V : la proaccélérine. On distingue la facteur V plaquettaire et le facteur V plasmatique. Le déficit en facteur V plasmatique est extrêmement rare et d'origine génétique, à transmission autosomale récessive. Chez les hétérozygotes (un seul gène muté) l'hémophilie est asymptomatique, alors que chez les homozygotes (les 2 chromosomes portent le gène muté), les hémorragies atteignent les tissus cutanéomuqueux. En ce qui concerne le facteur V plaquettaire ou facteur V Québec, son déficit provoque une maladie hémorragique extrêmement rare et sévère, qui se transmet de façon autosomique dominante. Le rôle essentiel du facteur V est d'accélérer la transformation de la prothrombine en thrombine. * Facteur VI : l'accélérine. Molécule proche de la précédente, qui intervient dans la transformation de la prothrombine en thrombine. * Facteur VII : la proconvertine (molécule très proche de la convertine). Son déficit est héréditaire provoque des hémorragies plus ou moins graves et se transmet de façon autosomale récessive, comme le facteur V. * Facteur VIII ou facteur antihémophilique A ou thromboplastinogène. Son déficit entraîne l'hémophilie A. C'est une glycoprotéine codée par un gène situé sur le bras long du chromosome X, qui a pu être cloné et donc synthétisé par génie génétique. Gros avantage : on produit un facteur VIII parfaitement pur et indemne de toute trace virale. Il est synthétisé dans le foie (par les hépatocytes), mais aussi dans d'autres organes comme la rate, le rein. En réalité, le facteur VIII est très instable et exposé à des réactions enzymatiques dès qu'il se retrouve dans la circulation sanguine. C'est la raison pour laquelle il s'associe au VWF (facteur de von Willebrand) et forme le complexe VIII. Cette liaison empêche entre autres, la protéolyse du facteur VIII. Dans la cascade de la coagulation, il joue un rôle fondamental en activant ou en catalysant des étapes de cette cascade, notamment pour les facteurs IX et X. * Facteur VWF ou facteur de von Willebrand : cette protéine est synthétisée par les cellules de l'endothélium vasculaire et par les mégacaryocytes (Dans notre sang, des éléments appelés plaquettes ou thrombocytes, jouent un rôle très important dens les mécanismes de la coagulation. Tout commence dans la moelle osseuse (organe hématopoïétique) où se trouve une lignée particulière de cellules : les mégacaryoblastes. Ce sont les précurseurs des thrombocytes. Elles se différencient progressivement en mégacaryocytes, très grosses cellules (plus de 30 µ) avec un énorme noyau multilobé. Ces cellules représentent un stade intermédiaire entre les mégacaryoblastes et les plaquettes ou thrombocytes.) Le facteur Willebrand assure plusieurs rôles très importants dans la coagulation : * il interviennent dans l'hémostase primaire en permettant l'activation et l'agrégation des plaquettes au niveau de la lésion vasculaire. * Il protège le facteur VIII instable en s'associant avec lui et en empêchant sa dégradation précoce. Son déficit provoque la maladie de Willebrand, au cours de laquelle on observe aussi une baisse du facteur VIII, du fait qu'il n'est plus protégé. * Facteur IX ou facteur antihémophilique B ou facteur Christmas ou PTC (Plasma Thromboplastin Component) : ce facteur intervient dans la voie intrinsèque et son déficit provoque l'hémophilie B. * Facteur X ou facteur Stuart ou facteur Prower : il agit dans les 2 voies (intrinsèque et extrinsèque) de la transformation de la prothrombine en thrombine. Le déficit en facteur X correspond généralement à une maladie génétique à transmission autosomique récessive et les hétérozygote (le gène muté n'est présent que sur un seul chromosome) sont en principe asymptomatiques. Chez les homozygotes, on a décrit des hémarthroses (hémorragies au niveau des articulations) et des hémorragies cérébroméningées. * Facteur XI ou facteur de Rosenthal ou PTA (Plasma Thromboplastin Antecedent) : ce facteur intervient dans la voie intrinsèque de la coagulation et son déficit provoque le syndrome de Rosenthal qui est un syndrome hémorragique. Il participe à la formation de la thrombine, produit impliqué à la fois dans la formation de la fibrine et la protection contre la fibrinolyse. * Facteur XII ou facteur Hageman (du nom du malade chez lequel il a été découvert) : c'est l'activation de ce facteur normalement présent dans le plasma, qui déclenche la voie intrinsèque de la coagulation. Le déficit en facteur XI provoque l'allongement du temps de coagulation, parfois des saignements (syndrome hémorragique). * Facteur XIII ou FSF (facteur stabilisant la fibrine) : son rôle est essentiel, car c'est lui qui permet la polymérisation des monomères en polymères de fibrine. Son déficit est rare et dû à une transmission génétique sur le mode autosomique récessif. Il provoque une forme d'hémophilie plus ou moins marquée chez les sujets homozygotes. III - Bilan sanguin : * bilan : de l'italien balancio, balance ; * sanguin : du latin sanguis, sanguinis [sanguin(o)-, -sanguin, -sanguinolent], liquide rouge qui circule dans les veines et les artères. Le bilan sanguin consiste à effectuer un certain nombre d'analyses, de dosages ou de recherches d'éléments particuliers dans le sang d'un patient. Le nombre de ces analyses est important et très variable selon la pathologie recherchée. Je ne citerai donc dans ce paragraphe que les actes les plus couramment demandés par le médecin prescripteur. Pour chacun de ces actes, des renseignements plus complets sont disponibles dans les pages de ce site en recherchant les différentes définitions. * Hémogramme, NFS (numération formule sanguine) : détermine le nombre ou la quantité de chacun des éléments figurés du sang : hématies ou globules rouges ou érythrocytes, leucocytes ou globules blancs (polynucléaires, lymphocytes, monocytes) et plaquettes ou thrombocytes. * VS ou vitesse de sédimentation : augmentée dans de nombreuses pathologies. * Ionogramme : dosage des différents ions présents dans le plasme : K+ (potassium), Na+ (sodium), Cl- (chlore) entre autres. * Glucose : la glycémie ou taux de glucose sanguin permet de dépister ou de suivre l'évolution d'un diabète. * Urée, créatinine ... On peut également cibler tel ou tel organe. Dans le bilan hépatique, on procède au dosage de : TxP, protéines, bilirubine, ASAT GO, ALAT GP, Gamma GT, PAL, alcoolémie ; dans le bilan lipidique, on évalue le cholestérol et les triglycérides etc. IV - Composition du sang : Le sang total (dont on n'a retiré aucun élément) est composé d'une phase liquide, le plasma et d'une phase solide : les cellules sanguines. * Le plasma : il contient essentiellement de l'eau,des ions (Na+, Cl-, K+, HCO3-), des nutriments provenant de la digestion, des protéines (albumine, facteurs de coagulation, immunoglobulines), des hormones, vitamines et déchets du métabolisme. Dans le plasma se trouve aussi une protéine en dissolution : le fibrinogène, capable de se polymériser en fibrine. Le sérum sanguin n'est autre que du plasma débarrassé de son fibrinogène. * Les cellules sanguines ou éléments figurés : - Les hématies : * hémato : du grec haima, [-émie, héma-, hémat(o)-, hémo-] : relatif au sang ; * ie : du suffixe -ie, -ié, -é qui transforme une racine ou un adjectif en un substantif. C'est le globule rouge ou érythrocyte, cellule anucléée (il n'y a plus de noyau) du sang. Il y en a environ cinq millions par mm3 de sang. Ces cellules ont une durée de vie courte (environ 120 jours), une dimension de 7 x 2 microns, et sont responsables du transport des gaz de la respiration : dioxygène et dioxyde de carbone, grâce à l'hémoglobine qu'elles transportent. Avec le monoxyde de carbone (CO) l'hémoglobine forme un composé stable responsable d'asphyxie et d'accidents mortels. A leur surface, des marqueurs dits "mineurs" déterminent les groupes sanguins et le facteur rhésus. Quelques renseignements supplémentaires sur l'hémoglobine : * hémo : du grec haima, [-émie, héma-, hémat(o)-, hémo-] : relatif au sang ; * globine : du latin globus [glob(o)-, -globine, globul(o)-, -globulie], sphérique, petit corps arrondi. L'hémoglobine est une protéine formée par quatre chaînes polypeptidiques deux à deux identiques et possédant chacune un groupement non protéique (c'est donc une hétéroprotéine) appelé hème et contenant du fer (ce qui explique que notre sang soit rouge). Son PM ou poids moléculaire est de 68 000. L'hémoglobine (symbole Hb) fixe de façon réversible à une, deux, trois ou quatre molécules de dioxygène O2. L'hémoglobine est contenue dans les globules rouges (ou hématies ou érythrocytes) et les mutations des gènes qui codent pour les molécules constituant cette hémoglobine, sont nombreuses et provoquent des maladies héréditaires telles la drépanocytose, les thalassémies, des hémophilies. Chez l'homme plusieurs hémoglobines se succèdent au cours de la vie et, à tout moment, il en existe plusieurs simultanément. Deux types de chaînes alpha et bêta sont présentes lors de la vie embryonnaire. Il s'agit : de la chaîne zêta (la première à apparaître), de la chaîne alpha proprement dite, de la chaîne epsilon (spécifique à la vie embryonnaire) et des chaînes gamma qui deviendront majoritaires chez le ftus. Six mois après la naissance le profil hémoglobinique de l'adulte est atteint : l'HbA1 ou hémoglobine A1 (2 chaînes alpha et 2 chaînes bêta - composée de 6 fractions : a, b, c, d, e et II) représente alors plus de 97 % de la totalité des hémoglobines. Une fraction de cette hémoglobine, appelée hémoglobine glyquée ou Hbg ou HbA1c (encore appelée hémoglobine glycosylée par certains laboratoires) présente une molécule de glucose à l'extrémité de chacune des chaînes ß. Il existe un autre constituant mineur, l'HbA2 ou hémoglobine A2 (2 chaînes alpha et 2 chaînes delta), exprimé à un taux d'environ 2,5 % dont la synthèse débute dans la période néonatale (chez le nouveau-né). L'HbF (ou hémoglobine F) quant à elle, n'existe plus qu'à l'état de traces inférieures à 1 %. Les gènes de la famille alpha sont situés dans la région distale du chromosome 16 et ceux de la famille bêta sur le chromosome 11. La famille alpha comporte trois gènes fonctionnels. La famille bêta en compte cinq : un gène embryonnaire (epsilon), deux gènes ftaux (gamma) et deux gènes adultes (bêta et delta). On connaît actuellement plusieurs centaines d'allèles des gènes codant pour l' aglobine ou pour la ßglobine : certains assez répandus, d'autres rarissimes. L'allèle considéré comme "normal" est celui que l'on rencontre le plus dans l'espèce humaine. Les mutations de ces gènes entraînent une diminution ou une absence de synthèse d'une ou plusieurs globines et de telles maladies imposent des transfusions répétées, avec une espérance de vie des malades qui ne dépasse que rarement la vingtaine d'années. Les athalassémies ou thalassémies a sont souvent causées par des délétions des gènes de l'aglobine (chaîne a) portés par la paire de chromosomes n° 16. Il en résulte la formation de deux hémoglobines pathologiques appelées HbH (hémoglobine H) et HbBart (hémoglobine Bart). Les ßthalassémies ou thalassémies ß, (ou maladie de COOLEY ou anémie de COOLEY) fréquemment causées par des mutations ponctuelles affectant les gènes de la ßglobine (chaîne ß) portés par la paire de chromosomes n° 11. Il existe une forme dite delta ou thalassémies d qui affecte l'HbA2 et qui passe inaperçue car asymptomatique. - Les leucocytes : * leuco : du grec leukos [leuc(o)-, leuk(o)-], blanc ; * cyte : du grec kutos [cyto-, -cyte, -cytie], cellule. Les leucocytes ou globules blancs ont un rôle essentiel dans les défenses de l'organisme : phagocytose et défense immunitaire à médiation cellulaire. certains d'entre eux ont des granulations dans le cytoplasme : ce sont les granulocytes ou polynucléaires (car on a cru longtemps qu'ils possédaient plusieurs noyaux - en fait il s'agit d'un seul noyau, multilobé). Dans ces granulocytes, on distingue essentiellement 3 catégories selon leur affinité pour les colorants : les basophiles qui se colorent préférentiellement avec des colorants basiques (pH > 7), les éosinophiles ou acidophiles qui réagissent aux colorants acides (pH < 7) et les neutrophiles qui se colorent avec des colorants neutres (pH = 7). Un deuxième groupe de leucocytes est représenté par agranulocytes, c'est-à-dire qu'ils n'ont pas de granulations dans leur cytoplasme. On y trouve les lymphocytes, petites cellules à gros noyau, avec les lymphocytes B qui acquièrent leur immunocompétence dans la moelle osseuse et qui, après transformation en plasmocytes, fabriquent les anticorps, et les lymphocytes T (ils acquièrent leur immunocompétence dans le thymus). Autre groupe dans les agranulocytes : les monocytes, grosses cellules à noyau excentré qui interviennent dans la phagocytose. Les valeurs normales oscillent entre 4000 et 10 000 leucocytes par mm3 de sang. Le comptage des leucocytes ou globules blancs est un leucogramme (du grec gramma, [-gramme], lettre, écriture et par extension, enregistrement écrit). - Les thrombocytes : * thrombo : du grec thrombos [thromb(o)-], caillot, en rapport avec la coagulation du sang ; * cyte : du grec kutos [cyto-, -cyte, -cytie], cellule. Le thrombocyte est l'un des éléments figurés du sang, plus connu sous le nom de plaquette (abréviation souvent utilisée : PLT).C'est le plus petit (2 à 4 µ) de tous ces éléments (les autres sont les globules rouges ou hématies ou érythrocytes et les globules blancs ou leucocytes) et en fait, ce thrombocyte n'est qu'un fragment d'une cellule initiale plus grande : le mégacaryocyte, situé dans la moelle osseuse. La durée de vie des plaquettes est environ comprise entre 7 et 10 jours, c'est-à-dire que, passé ce délai, toutes les plaquettes ont été détruites par la rate et remplacées par de nouvelles. Elles on un rôle déterminant dans la coagulation du sang pour arrêter un saignement (c'est l'hémostase) et leur absence ou leur insuffisance peut être une cause majeure d'hémophilie. La thrombocytémie (du grec haima, [-émie, héma-, hémat(o)-, hémo-] : relatif au sang) est le nombre de plaquettes dans le sang. Les valeurs normales sont comprises entre 150 000 et 450 000 par mm3 de sang. Un examen appelé "temps de saignement" (TS) permet d'évaluer l'état fonctionnel des thrombocytes. V - Histocompatibilité : * histo : du grec histos ou histion [hist(o), histio-], tissu ; * compatibilité : du latin compati [-compatibilité], souffrir avec. Similitude des antigènes codés par le CMH (complexe majeur d'histocompatibilité) entre le donneur et le receveur, nécessaire en cas de greffe.Ces antigènes sont présents sur toutes nos cellules à noyau. Sur les hématies (cellules sans noyau) se trouvent des antigènes qui déterminent les groupes sanguins et qui déterminent le complexe mineur d'histocompatibilité.
© Georges Dolisi | |


| < vers le menu principal | Politique de confidentialité | App Privacy Policy | ^ haut de la page |
| © 2006, 2020 Medicopedia | Powered by JIMaroc |