| Dictionnaire médical |
Abrév Abréviations H Hall Hanc Hapt Hept Hern Herp Hexa Hexo Hidr Hist Histo Holo Holt Homi Homme Homo Homé Homéo Horm Hormon Hume Humé Humér Hunt Hyal Hyda Hydat Hydr Hydro Hydrox Hygi Hype Hyper Hyph Hypn Hypo Hypé Hyst Hystér Hédo Héli Hém Héma Hémato Hémi Hémo Hépa Hépat Hépato Héro Hété Hétéro
| Hémoglobine Bart | |
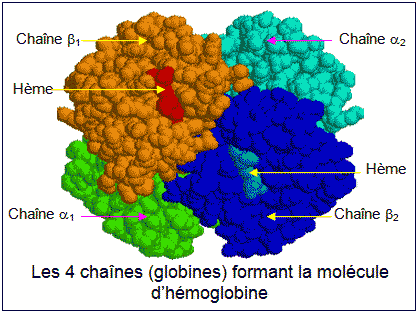
| Hématologie, génétique N. f. * hémo : du grec haima, {-émie, héma-, hémat(o)-, hémo-} : relatif au sang ; * globine : du latin globus {glob(o)-, -globine, globul(o)-, -globulie}, sphérique, petit corps arrondi. L'hémoglobine est une protéine formée par quatre chaînes polypeptidiques deux à deux identiques et possédant chacune un groupement non protéique (c'est donc une hétéroprotéine) appelé hème et contenant du fer (ce qui explique que notre sang soit rouge). Son PM ou poids moléculaire est de 68 000. L'hémoglobine (symbole Hb) fixe de façon réversible à une, deux, trois ou quatre molécules de dioxygène O2. L'hémoglobine est contenue dans les globules rouges (ou hématies ou érythrocytes) et les mutations des gènes qui codent pour les molécules constituant cette hémoglobine, sont nombreuses et provoquent des maladies héréditaires telles la drépanocytose, les thalassémies, des hémophilies. Chez l'homme plusieurs hémoglobines se succèdent au cours de la vie et, à tout moment, il en existe plusieurs simultanément. Deux types de chaînes alpha et bêta sont présentes lors de la vie embryonnaire. Il s'agit : de la chaîne zêta (la première à apparaître), de la chaîne alpha proprement dite, de la chaîne epsilon (spécifique à la vie embryonnaire) et des chaînes gamma qui deviendront majoritaires chez le ftus. Six mois après la naissance le profil hémoglobinique de l'adulte est atteint : l'HbA1 ou hémoglobine A1 (2 chaînes alpha et 2 chaînes bêta - composée de 6 fractions : a, b, c, d, e et II) représente alors plus de 97 % de la totalité des hémoglobines. Il existe un autre constituant mineur, l'HbA2 ou hémoglobine A2 (2 chaînes alpha et 2 chaînes delta), exprimé à un taux d'environ 2,5 % dont la synthèse débute dans la période néonatale (chez le nouveau-né). L'HbF (ou hémoglobine F) quant à elle, n'existe plus qu'à l'état de traces inférieures à 1 %. Les gènes de la famille alpha sont situés dans la région distale du chromosome 16 et ceux de la famille bêta sur le chromosome 11. La famille alpha comporte trois gènes fonctionnels. La famille bêta en compte cinq : un gène embryonnaire (epsilon), deux gènes ftaux (gamma) et deux gènes adultes (bêta et delta). On connaît actuellement plusieurs centaines d'allèles des gènes codant pour l' aglobine ou pour la ßglobine : certains assez répandus, d'autres rarissimes. L'allèle considéré comme "normal" est celui que l'on rencontre le plus dans l'espèce humaine. Les mutations de ces gènes entraînent une diminution ou une absence de synthèse d'une ou plusieurs globines et de telles maladies imposent des transfusions répétées, avec une espérance de vie des malades qui ne dépasse que rarement la vingtaine d'années. Les athalassémies ou thalassémies a sont souvent causées par des délétions des gènes de l'aglobine (chaîne a) portés par la paire de chromosomes n° 16. Il en résulte la formation de deux hémoglobines pathologiques appelées HbH (hémoglobine H) et HbBart (hémoglobine Bart). Les ßthalassémies ou thalassémies ß, (ou maladie de COOLEY ou anémie de COOLEY) fréquemment causées par des mutations ponctuelles affectant les gènes de la ßglobine (chaîne ß) portés par la paire de chromosomes n° 11. Il existe une forme dite delta ou thalassémies d qui affecte l'HbA2 et qui passe inaperçue car asymptomatique.
© Georges Dolisi | |
| < vers le menu principal | Politique de confidentialité | App Privacy Policy | ^ haut de la page |
| © 2006, 2020 Medicopedia | Powered by JIMaroc |