| Dictionnaire médical |
Abrév Abréviations C Cach Caco Cadh Cadu Caec Cail Cal Calc Calcan Calcul Call Camp Campt Cana Canc Cancer Cand Capi Capill Caps Carb Carc Carcin Card Cardi Cardio Carm Cart Cary Carè Caré Cata Cath Cato Caté Caus Cave Cavi Cavo Cavu Cein Cell Cellul Cent Cerv Cervic Cham Chei Chev Chik Chim Chimio Chir Chla Chlo Chlor Chol Chon Chondr Chor Chori Choré Chro Chrom Chroma Chromo Chry Chyl Chym Chéi Chéil Chél Chém Cinq Circ Cirr Cirs Clai Clar Clau Clav Clea Clim Clin Clit Clon Cléid Coag Coar Cobl Cocc Coch Cochlé Coda Code Codo Codé Coeli Col Coli Coll Colo Colp Coma Comp Comé Con Conc Cond Cong Coni Conj Conn Cons Cont Contra Contre Conv Coor Coph Copr Coqu Cord Corn Coro Corp Cort Cortic Cory Cost Coty Cox Coxa Coxo Cran Crani Cras Craw Cres Crev Cric Cris Crow Crue Cruo Crur Crya Crye Cryo Cryp Crypto Crân Créa Créat Crén Crép Cubi Cubit Cura Curc Cure Curi Cut Cyan Cycl Cyph Cyst Cyth Cyto Cli Cur Cæca Cæco Cæcu Céph Céphal Céré Cérébe Cérébr Côlo Côn Cône Côte
| Coagulation sanguine | |
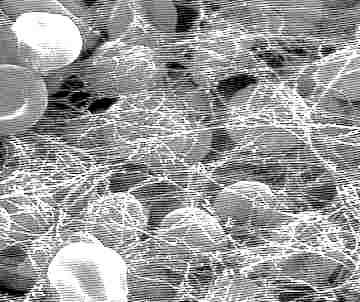
| Endocrinologie et métabolismes, hématologie, médecine biologique N. f. * coagulation : du latin coagulare {-coagulant, -coagulation}, donner à un liquide une consistance solide, le figer ; * sanguine : du latin sanguis, sanguinis, sanguinare {sanguin(o)-, -sanguin, -sanguinolent}, liquide rouge qui circule dans les veines et les artères. A l'exception du facteur IV qui est le calcium (Ca++), les facteurs de coagulation sont toutes des protéines, substances présentes dans le sang et qui participent à la coagulation sanguine ou hémostase. Toute anomalie ou dysfonctionnement d'un ou plusieurs de ces facteurs provoque une forme plus ou moins sévère d'hémophilie. On connaît actuellement 13 facteurs de coagulation qui sont notés par des chiffres romains. Pour mieux comprendre ce qui suit, petit rappel sur la coagulation. C'est un ensemble de mécanismes qui fait passer le sang de l'état liquide à un état de gel ou caillot, en transformant le fibrinogène soluble en fibrine insoluble qui forme l'armature du caillot. On distingue classiquement 3 étapes : 1) La thromboplastinoformation ou transformation de la prothrombinase selon 2 voies différentes : intrinsèque et extrinsèque, et qui aboutit à la formation d'une enzyme : le facteur X activé. 2) La thrombinoformation qui aboutit à la formation de thrombine sous l'action de la thrombinase. 3) La fibrinoformation qui permet, grâce à la thrombine, la transformation du fibrinogène en fibrine. * Facteur I : c'est le fibrinogène. Du latin et du grec genesis [-gène, -genèse, génique], naissance, formation, qui engendre. Synthétisé par le foie, le fibrinogène est une glycoprotéine qui existe à l'état soluble dans le sang circulant. Sous l'action d'une enzyme (la thrombine), le fibrinogène est transformé en fibrine insoluble (c'est la fibrino-formation) et joue donc un rôle majeur dans la coagulation du sang (hémostase). A l'électrophorèse, le fibrinogène migre entre les ß et les g globulines. Les valeurs plasmatiques normales sont comprises entre 3 à 5 g/L (ou 9 à 15 mmol/L) de fibrinogène par litre de sang. C'est la fibrinogénémie, alors que la fibrinémie représente le taux de fibrine du plasma. * Facteur II : la prothrombine. Du grec thrombos [thromb(o)-], caillot, en rapport avec la coagulation du sang. C'est une substance qui, sous l'influence d'une dizaine d'autres facteurs, fait coaguler le sang par polymérisation du fibrinogène soluble en fibrine insoluble. La mesure de la prothrombine est très importante chez les personnes qui suivent un traitement par anticoagulants. On pratique essentiellement 2 examens : 1 - Le taux de prothrombine permet de tester le bon fonctionnement de certains facteurs de la coagulation. Ils sont abaissés en cas de maladie hépatique et lorsqu'un malade prend des médicaments anticoagulants par voie orale. C'est l'examen de choix pour contrôler l'efficacité d'un traitement. Ce taux de prothrombine ou "TP", doit alors être strictement compris entre 25% et 35%, alors qu'il est normalement supérieur à 70%. S'il est inférieur à 25%, il y a risque d'hémorragie, et s'il est supérieur à 35%, le traitement n'est plus suffisamment efficace. 2 - Le temps de prothrombine, appelé également temps de Quick, est le temps que met le plasma sanguin (partie liquidienne du sang) à coaguler quand on le met en présence dun extrait de tissu humain, animal ou synthétique appelé thromboplastine, cytozyme ou thrombokinase. Il sagit dun ensemble denzymes nécessaires à la coagulation du sang et permettant de transformer la prothrombine en thrombine. La mesure du temps de Quick est faite à partir dun prélèvement de sang du malade. Ce test sert à savoir si le patient présente une tendance à faire des hémorragies à la suite dune carence en certains facteurs normalement présents dans le sang : facteur II, V, VII, X qui interviennent dans la coagulation normale du sang. Le temps de prothrombine sexprime sous la forme dun indice INR (International Normalized Ratio ou rapport international normalisé). Un temps de prothrombine anormal indique un problème de coagulation, dorigine soit pathologique soit thérapeutique. Une atteinte du foie ou un manque de vitamine K peuvent être à lorigine de perturbations du temps de prothrombine. Le TP sert à surveiller les traitements anticoagulants par les antivitamines K. On appelle facteur IIa la thrombine. * Facteur III ou facteur tissulaire : la thromboplastine tissulaire ou thrombokinase (les autres sont des facteurs plasmatiques). Ce facteur III de la coagulation est présent à la surface des vaisseaux sanguins. Sa quantité à la surface augmente selon certains signaux comme l'inflammation. Elle provoque l'activation du facteur VII de la coagulation - voir ci-dessous. * Facteur IV : le calcium. Du latin calx, calcis, chaux. Métal de symbole chimique Ca. C'est l'élément N° 20 de masse atomique Ca = 40,08 et de densité 1,54. Il s'oxyde à l'air en formant la chaux vive CaO. Il participe à un grand nombre de phénomènes physiologiques : synthèse du tissu osseux et dentaire, coagulation du sang, conduction nerveuse, contraction musculaire. L'hypocalcémie provoque des convulsions et des tremblements (spasmophilie, tétanie). * Facteur V : la proaccélérine. On distingue la facteur V plaquettaire et le facteur V plasmatique. Le déficit en facteur V plasmatique est extrêmement rare et d'origine génétique, à transmission autosomale récessive. Chez les hétérozygotes (un seul gène muté) l'hémophilie est asymptomatique, alors que chez les homozygotes (les 2 chromosomes portent le gène muté), les hémorragies atteignent les tissus cutanéomuqueux. En ce qui concerne le facteur V plaquettaire ou facteur V Québec, son déficit provoque une maladie hémorragique extrêmement rare et sévère, qui se transmet de façon autosomique dominante. Le rôle essentiel du facteur V est d'accélérer la transformation de la prothrombine en thrombine. * Facteur VI : l'accélérine. Molécule proche de la précédente, qui intervient dans la transformation de la prothrombine en thrombine. * Facteur VII : la proconvertine (molécule très proche de la convertine). Son déficit est héréditaire provoque des hémorragies plus ou moins graves et se transmet de façon autosomale récessive, comme le facteur V. * Facteur VIII ou facteur antihémophilique A ou thromboplastinogène. Son déficit entraîne l'hémophilie A. C'est une glycoprotéine codée par un gène situé sur le bras long du chromosome X, qui a pu être cloné et donc synthétisé par génie génétique. Gros avantage : on produit un facteur VIII parfaitement pur et indemne de toute trace virale. Il est synthétisé dans le foie (par les hépatocytes), mais aussi dans d'autres organes comme la rate, le rein. En réalité, le facteur VIII est très instable et exposé à des réactions enzymatiques dès qu'il se retrouve dans la circulation sanguine. C'est la raison pour laquelle il s'associe au VWF (facteur de von Willebrand) et forme le complexe VIII. Cette liaison empêche entre autres, la protéolyse du facteur VIII. Dans la cascade de la coagulation, il joue un rôle fondamental en activant ou en catalysant des étapes de cette cascade, notamment pour les facteurs IX et X. * Facteur VWF ou facteur de von Willebrand : cette protéine est synthétisée par les cellules de l'endothélium vasculaire et par les mégacaryocytes (Dans notre sang, des éléments appelés plaquettes ou thrombocytes, jouent un rôle très important dens les mécanismes de la coagulation. Tout commence dans la moelle osseuse (organe hématopoïétique) où se trouve une lignée particulière de cellules : les mégacaryoblastes. Ce sont les précurseurs des thrombocytes. Elles se différencient progressivement en mégacaryocytes, très grosses cellules (plus de 30 µ) avec un énorme noyau multilobé. Ces cellules représentent un stade intermédiaire entre les mégacaryoblastes et les plaquettes ou thrombocytes.) Le facteur Willebrand assure plusieurs rôles très importants dans la coagulation : * il interviennent dans l'hémostase primaire en permettant l'activation et l'agrégation des plaquettes au niveau de la lésion vasculaire. * Il protège le facteur VIII instable en s'associant avec lui et en empêchant sa dégradation précoce. Son déficit provoque la maladie de Willebrand, au cours de laquelle on observe aussi une baisse du facteur VIII, du fait qu'il n'est plus protégé. * Facteur IX ou facteur antihémophilique B ou facteur Christmas ou PTC (Plasma Thromboplastin Component) : ce facteur intervient dans la voie intrinsèque et son déficit provoque l'hémophilie B. * Facteur X ou facteur Stuart ou facteur Prower : il agit dans les 2 voies (intrinsèque et extrinsèque) de la transformation de la prothrombine en thrombine. Le déficit en facteur X correspond généralement à une maladie génétique à transmission autosomique récessive et les hétérozygote (le gène muté n'est présent que sur un seul chromosome) sont en principe asymptomatiques. Chez les homozygotes, on a décrit des hémarthroses (hémorragies au niveau des articulations) et des hémorragies cérébroméningées. * Facteur XI ou facteur de Rosenthal ou PTA (Plasma Thromboplastin Antecedent) : ce facteur intervient dans la voie intrinsèque de la coagulation et son déficit provoque le syndrome de Rosenthal qui est un syndrome hémorragique. Il participe à la formation de la thrombine, produit impliqué à la fois dans la formation de la fibrine et la protection contre la fibrinolyse. * Facteur XII ou facteur Hageman (du nom du malade chez lequel il a été découvert) : c'est l'activation de ce facteur normalement présent dans le plasma, qui déclenche la voie intrinsèque de la coagulation. Le déficit en facteur XI provoque l'allongement du temps de coagulation, parfois des saignements (syndrome hémorragique). * Facteur XIII ou FSF (facteur stabilisant la fibrine) : son rôle est essentiel, car c'est lui qui permet la polymérisation des monomères en polymères de fibrine. Son déficit est rare et dû à une transmission génétique sur le mode autosomique récessif. Il provoque une forme d'hémophilie plus ou moins marquée chez les sujets homozygotes.
© Georges Dolisi | |
| < vers le menu principal | Politique de confidentialité | App Privacy Policy | ^ haut de la page |
| © 2006, 2020 Medicopedia | Powered by JIMaroc |