| Dictionnaire médical |
A Ab Aba Abcè Abcé Abdo Abdomi Abdu Abio Ablu Abor Abou Abra Abru Abréviations Abso Aca Acan Acanth Acar Acat Ace Acep Acet Ach Ache Achi Achl Achlor Acho Achr Achro Aché Acid Acin Acné Acor Acou Acro Acti Actin Actino Acu Acui Acum Acup Acya Acyl Acys Acép Acét Adac Addi Addu Adia Adip Adré Adrén Adyn Adén Affe Affé Agal Agas Aggl Agglut Agna Agno Agom Agor Agra Agry Agén Aine Aire Akin Alal Albi Albu Alca Alco Alcool Aldo Aldé Alec Aleu Alex Alg Algh Algi Algo Algé Alit Alle Allerg Allo Allè Allé Alog Alop Alph Alpha Alvé Alvéol Alym Amar Amau Amb Ambi Ambl Ambly Ami Amib Amid Amido Amim Amin Amit Ammo Amni Amné Amon Amor Amph Amphi Ampho Ampo Ampu Ampull Amy Amyc Amyg Amygda Amyl Amyo Amyx Amyè Amyé Amb Amèl Amél Amét An Ana Anab Anad Anal Anam Anap Anas Anat Anaé Andr Ane Anen Aner Anes Aneu Ang Angi Angin Ango Angé Anh Anhi Anhy Anhé Anis Anit Anky Ankyl Anne Annu Anod Anom Anop Anor Anos Anot Anov Anox Ant Anta Anth Anthra Anthro Anti Antr Anté Anur Anus Anxi Ans Ané Anéc Aném Anér Anév Anévri Aort Ap Apat Apep Apex Aph Apha Aphi Apho Apht Aphy Apic Apip Apl Apla Apn Apo Apod Apol Apon Apop Appa Appe Append Appé Apr Apra Apro Apy Apyr Aqua Aque Arac Arachn Arch Arno Arom Arth Arthr Artè Arté Artéri Ary Aryt Arytén Aréf Aréo Asci Asep Asex Asia Aspe Asph Asta Asth Asthm Asti Astr Astro Asym Asyn Asys Atax Athr Athy Athé Athér Aton Atop Atri Atro Atré Atti Attic Atyp Atél Audi Auri Auti Auto Avol Avul Ax Axia Axil Axis Axon Axoï Azog Azop Azor Azos Azot Azym Aér Aéra Aéro alph
| Analphalipoprotéinémie | |
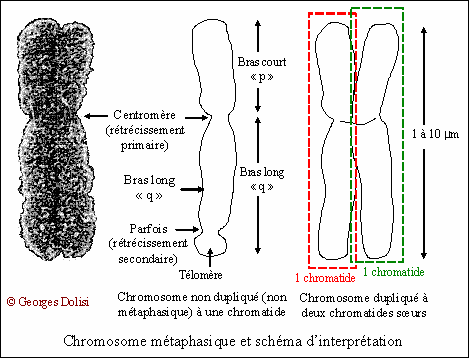
| Génétique, maladies orphelines, hépatogastroentérologie N. f. * Tangier : du nom d'une ville sur l'île "Tangier Island" dans la Baie de Chesapeake, au large de la Virginie ; * hypo : du grec hupo , dessous, indiquant aussi une qualité ou une intensité inférieures à la normale ; * alpha : première lettre de lalphabet grec (a), utilisée avec dautres lettres grecques pour classer des éléments en biochimie, ou pour désigner un rayonnement en physique ou en médecine nucléaire ; * lipo : du grec lipos, liparos {lip(o)-}, graisse, gras ; * protéine : du grec tardif prôteios, signifiant "qui occupe le premier rang" ou "de première qualité", lui-même dérivé de protos, premier, relatif aux protides ou aux protéines, composées essentiellement de C, H, O et N ; * émie : du grec haima, {-émie, héma-, hémat(o)-, hém(o)-} : relatif au sang. [Angl. : Tangier disease, analphalipoproteinemia, hypoalphalipoproteinemia] Syn. : hypoalphalipoprotéinémie, analphalipoprotéinémie, syndrome de Tangier. La maladie de Tangier est une maladie génétique rare ou maladie orpheline, puisqu'on ne connaît que moins de cinquante cas dans une vingtaine de familles dans le monde. Quelques explications pour mieux comprendre cette maladie du transport du cholestérol. * Sur le chromosome 9q31, c'est-à-dire sur le bras long (q) du 9e chromosome, à un emplacement ou locus précis nommé 31, se trouve le gène ABCA1 (ATP Binding Cassette Transporter). En fait, il s'agit de la 9e paire de chromosomes, et nous avons donc ce gène en double, chacun hérité de l'un de nos deux parents. ATP désigne l'adénosine triphosphate qui est le principal transporteur d'énergie dans toutes nos réactions cellulaires. Ce gène ABCA1 code la fabrication d'une protéine du même nom (ABCA1) qui est enchâssée dans la membrane de nos cellules et dont le rôle principal est de permettre à certaines molécules dont le cholestérol, de quitter l'intérieur de la cellule pour passer dans le milieu extracellulaire. Ces molécules sont trop grandes pour passer "naturellement" de l'intérieur vers l'extérieur et ce passage nécessite la présence dans la membrane de ces protéines particulières qui vont permettre ce transfert qualifié d'actif, en consommant de l'énergie, donc de l'ATP. Ce n'est qu'une fois dans le milieu extracellulaire que le cholestérol va se lier à l'apoA1 et aux phospholipides pour former le complexe HDL. * Chez un nombre très limité de personnes, l'un des chromosomes n° 9, plus rarement encore les deux, présente(nt) un gène ABCA1 qui a muté, c'est-à-dire qu'il ne fabrique plus la bonne protéine. Sans trop entrer dans les détails, on constate que dans un gène muté, la séquence des bases azotées qui composent la double hélice de l'ADN n'est plus la bonne. On comprend aisément ce qui se passe dans ce cas : le cholestérol ne peut plus quitter la cellule pour se rendre vers le foie et être éliminé. Il va s'accumuler préférentiellement dans des organes comme le foie et la rate, les artères, la cornée, les amygdales, entre autres. La maladie de Tangier résulte donc d'un dysfonctionnement du transport du cholestérol. * Une précision importante : Cette maladie se transmet selon le mode autosomique récessif. Autosomique : qualifie les chromosomes non sexuels, c'est-à-dire les 22 premières paires (la 23e paire étant conventionnellement celle des chromosomes sexuels, XX chez la femme, XY chez l'homme). Récessif : il faut que l'enfant reçoive de chacun de ses parents un chromosome avec le gène muté pour qu'il développe la maladie. Il est alors homozygote pour ce gène muté, puisqu'il l'a sur les deux chromosomes n° 9. S'il ne reçoit le gène muté que de l'un de ses parents, il est hétérozygote ou "porteur sain" et ne développe aucun symptôme de la maladie, car il produit suffisamment de protéines ABCA1 normales. * Principaux symptômes de la maladie de Tangier, sachant qu'aucun d'entre eux n'est obligatoire et qu'il existe des cas totalement asymptomatiques. ● Chez les nouveau-nés, les amygdales sont jaune-orangé du fait de l'accumulation d'esters du cholestérol et de taille supérieure à la normale. ● Hypolipidémie, en particulier baisse sévère du taux des HDL (high density lipoprotein, le "bon" cholestérol) qui est < 0,1 g/L, ainsi que de l'ApoA1. C'est ce qui justifie son autre nom d'hypoalphalipoprotéinémie ou d'analphalipoprotéinémie. ● Athérosclérose prématurée disséminée, avec atteinte coronarienne fréquente et risque accru d'infarctus du myocarde. ● Opacité cornéenne résultant de l'accumulation d'esters de cholestérol. ● Hépatomégalie et splénomégalie (augmentation anormale du volume du foie et de la rate). ● Sur la muqueuse du côlon sigmoïde et du rectum, on observe des dépôts jaunâtres de cholestérol, visibles en coloscopie sous forme de plaques de 2 à 3 mm. ● Les amygdales ne sont pas les seuls organes lymphoïdes touchés : on peut aussi observer de petites adénopathies inguinales (à l'aine), axillaires (aux aisselles) et cervicales. ● Le fait que le cholestérol ne puisse plus sortir des cellules va provoquer une cytolyse (destruction cellulaire) plus ou moins importante, ce qui se traduit par un taux d'ALAT supérieur à 50 UI/L (normale < 25). ● Parfois rétinite pigmentaire, légère hypertriglycéridémie, entre autres. * Traitements. Comme indiqué plus haut, il s'agit d'une maladie orpheline et il n'existe aujourd'hui aucun traitement, sinon symptomatique. Cela signifie que l'on soigne au coup par coup les symptômes qui deviennent gênants ou dangereux : angioplastie coronaire transluminale si des athéromes obstruent les artères coronaires, greffe de cornée pour éviter la cécité, amygdalectomie (ou tonsillectomie) chez les jeunes enfants, greffe du foie, entre autres. Les espoirs actuels reposent sur une thérapie génique qui n'est pas encore au point.
© Georges Dolisi | |
| < vers le menu principal | Politique de confidentialité | App Privacy Policy | ^ haut de la page |
| © 2006, 2020 Medicopedia | Powered by JIMaroc |